



بیماری شارکو ماری توث ؛ علائم ، تشخیص و درمان | کافه پزشکی
بیماری CMT یا شارکو ماری توث یک گروه از اختلالات است که در آن اعصاب محیطی حرکتی و یا حسی تحت تاثیر قرار می گیرند و موجب ضعف عضلانی و آتروفی و همچنین از دست دادن حس می شود . این تظاهرات ابتدا در پاها و بعدا در دست رخ می دهد. سلول های عصبی در افراد مبتلا به این اختلال قادر به ارسال سیگنال های الکتریکی به علت ناهنجاری های عصبی یا ناهنجاری های میلین اطراف آکسون نیستند. جهش های ژن خاص مسئول عملکرد غیر طبیعی اعصاب محیطی هستند. همراه کافه پزشکی باشید
آنچه در این پست کافه پزشکی خواهید دید
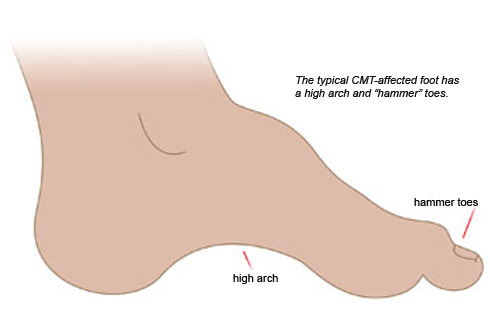
علائم و نشانه های بیماری شارکو ماری توث
علائم بیماری CMT معمولا به تدریج در نوجوانی آغاز می شود ، اما می تواند زودتر یا دیرتر شروع شود . تقریبا در تمام موارد، طولانی ترین الیاف عصبی ابتدا تحت تاثیر قرار می گیرند. پرچم های قرمز رایج ممکن است شامل کاهش حساسیت به گرما، لمس یا درد، ضعف عضلانی در دست و پا ، مشکل با مهارت های حرکتی، از دست دادن توده عضلانی در پا ، مکرر افتادن و پاهای صاف. رفلکس کششی ممکن است از دست رفته باشد. بیماری به آرامی پیشرونده و متغیر است و افراد مبتلا ممکن است عمر طبیعی خود را حفظ کنند. در موارد شدید، مشکلات تنفسی می توانند مرگ را تشدید کنند.
علل بیماری شارکو ماری توث
بیماری CMT می تواند در شکل اتوزومی غالب، اتوزوم مغلوب یا X-linked غالب به ارث برده شود.
اختلالات ژنتیکی پس از زایمان زمانی اتفاق می افتد که یک فرد همان ژن غیر طبیعی را برای همان صفات از هر یک از والدین به ارث برساند. اگر یک فرد یک ژن طبیعی و یک ژن برای این بیماری را دریافت کند، فرد برای این بیماری حامل است ، اما معمولا نشانه ها را نشان نمی دهد. خطر ابتلای کودک زمانی که دو والد حامل به هر دوی این ژن های معیوب باشند ، ۲۵٪ است. خطر حامل شدن کودک مانند پدر و مادر ، با هر حاملگی ۵۰٪ است. شانس یک کودک برای دریافت ژنهای نرمال از هر دو والدین و ژنتیکی طبیعی برای این ویژگی خاص ۲۵٪ است. این خطر برای مردان و زنان یکسان است.
اختلالات ژنتیکی غالب در زمانی رخ می دهد که تنها یک نسخه از یک ژن غیر طبیعی برای ظهور بیماری ضروری است. ژن غیر طبیعی ممکن است از هر دو والد به ارث برده شود یا می تواند یک جهش جدید (تغییر ژن) در فرد مبتلا باشد. خطر انتقال ژن غیرطبیعی از والدین آسیب دیده به فرزندان، ۵۰٪ برای هر بارداری صرف نظر از جنس فرزند حاصل می شود.
اختلالات ژنتیکی غالب وابسته به X مرتبط با یک ژن غیر طبیعی در کروموزوم X ایجاد می شود. زنان با یک ژن غیر طبیعی تحت بیماری قرار دارند. مردان با یک ژن غیر طبیعی بیش از زنان بیشتر تحت تاثیر قرار می گیرند.
نوروپاتی ارثی CMT به چندین نوع CMT1، CMT2، CMT3، CMT4 و CMTX تقسیم می شود.
CMT1 شکل غالب این وضعیت است که سرعت هدایت عصبی آن آهسته است و بسیار شایع تر از CMT2 است . CMT1 ناشی از ژن های غیر طبیعی درگیر در ساختار و عملکرد میلین است. CMT1 بیشتر به CMT1A، CMT1B، CMT1C، CMT1D و CMT1X تقسیم شده است، براساس اختلالات ژن خاص. CMT1A بوسیله تکثیر ژن PMP22 که در کروموزوم ۱۷ در ۱۷p11.2 واقع شده است. CMT1A شایع ترین نوع CMT1 است. CMT1B ناشی از اختلال در ژن MPZ واقع در کروموزوم ۱ در ۱q22 است. CMT1C ناشی از یک اختلال در ژن SIMPLE واقع در کروموزوم ۱۶ در ۱۶p13.1-p12.3 است. CMT1D ناشی از اختلال در ژن EGR2 واقع در کروموزوم ۱۰ در ۱۰q21.1-q22.1 است. CMT1X ناشی از جهش در GJB1 (واقع در Xq13.1)، ژن که پروتئین اتصال اتصال شکاف کدون ۳۲ را کدگذاری می کند. زیر گونه های کمتری از CMT1 هنوز ممکن است یافت شود.
CMT2 یک شکل غالب اتوزومی مغزی است که سرعت هدایت عصب معمولا طبیعی یا کمی کندتر از حد طبیعی است. CMT2 بوسیله ژن های غیر طبیعی درگیر در ساختار و عملکرد آکسون ها ایجاد می شود. CMT2 بیشتر به CMT2A – 2L تقسیم شده است بر اساس جهش در ژن های خاص. CMT2A، شایع ترین و ناشی از جهش در ژن MFN2 واقع در کروموزوم ۱ در ۱p36.2 است. CMT2B ناشی از جهش در ژن RAB7 واقع در کروموزوم ۳ در ۳q21 است. CMT2C بوسیله یک ژن ناشناخته در کروموزوم ۱۲ در ۱۲q23-34 ایجاد می شود. CMT2D ناشی از جهش در ژن GARS واقع در کروموزوم ۷ در ۷p15 است. CMT2E ناشی از جهش در ژن NEFL واقع در کروموزوم ۸ در ۸p21 است. CMT2F ناشی از جهش در ژن HSPB1 است. CMT2L ناشی از جهش در ژن HSPB8 است.
CMT3، همچنین به نام Dejerine-Sottas نامیده می شود، دیگر نامگذاری ژنتیکی مفید نیست، زیرا افراد مبتلا به این بیماری جهش ژنی را در یکی از ژن های مسئول CMT1A، CMT1B، CMT1D یا CMT4 یافت می شوند.
CMT4 یک فرم اتوزومی مغلوب از وضعیت است. این بیشتر به CMT4A، CMT4B1، CMT4B2، CMT4C، CMT4D، CMT4E و CMT4F تقسیم شده است. CMT4A ناشی از اختلال در ژن GDAP1 واقع در کروموزوم ۸ در ۸q13-q21 است. CMT4B1 ناشی از اختلال در ژن MTMR2 واقع در کروموزوم ۱۱ در ۱۱q22 است. CMT4B2 ناشی از اختلال در ژن SBF2 / MTMR13 واقع در کروموزوم ۱۱ در ۱۱p15 است. CMT4C ناشی از یک اختلال در ژن KIAA1985 واقع در کروموزوم ۵ در ۵q32 است. CMT4D ناشی از اختلال در ژن NDRG1 واقع در کروموزوم ۸ در ۸q24.3 است. CMT4E، همچنین به عنوان neuropathy hypomyelination مادرزادی شناخته می شود، ناشی از اختلال در ژن EGR2 واقع در کروموزوم ۱۰ در ۱۰q21.1-q22.1 است. CMT4F ناشی از اختلال در ژن PRX واقع در کروموزوم ۱۹ در ۱۹q13.1-q13.2 است. CMT4H ناشی از اختلال در ژن FDG4 است. CMT4J ناشی از اختلال در ژن FIG4 است. با این حال، اکثر موارد CMT2 ناشی از جهش در این ژنها نیست، به طوری که بسیاری از علل ژنتیکی هنوز کشف شده اند.
شیوع شارکو ماری توث
نشانه های نوروپاتی ارثی CMT معمولا به تدریج در نوجوانی یا سنین میانی شروع می شود. این وضعیت تعداد معینی از مردان و زنان را تحت تاثیر قرار می دهد. نوروپاتی ارثی CMT شایع ترین اختلال عصبی ارثی است که بیش از ۲۵۰،۰۰۰ آمریکایی را تحت تاثیر قرار می دهد. از آنجا که این بیماری غالبا تشخیص داده نشده است، تعداد واقعی افراد مبتلا ممکن است بالاتر باشد.
تشخیص شارکو ماری توث
تشخیص نوروپاتی ارثی CMT می تواند چالش برانگیز باشد. تشخیص بر اساس علائم جسمی، سابقه خانوادگی و آزمایشهای بالینی است. تست های بالینی شامل سرعت هدایت عصبی (NCV) است . آزمایش ژنتیکی مولکولی در حال حاضر برای CMT1A، CMT1B، CMT1D، CMT2E، CMT4A، CMT4E، CMT4F و CMTX در دسترس است.
درمان شارکو ماری توث
درمان نوروپاتی ارثی CMT علامتدار و حمایتی است. یک درمان قطعی در دسترس نیست، بنابراین علائم را به حداقل می رساند یا خاتمه می دهد. درمان های جامع شامل فیزیوتراپی، ارتوپدی و جراحی برای اصلاح عیوب . درمان های تکمیلی ممکن است به روانشناسی کمک کند، درد و ناراحتی را کاهش دهد و کیفیت کلی زندگی را بهبود بخشد. مشاوره حرفه ای، پیش بینی پیشرفت اختلال، ممکن است برای بیماران جوان مفید باشد.
منبع : rarediseases.org | ترجمه کافه پزشکی