

سندروم شارژه ؛ علائم ، تشخیص و درمان | کافه پزشکی
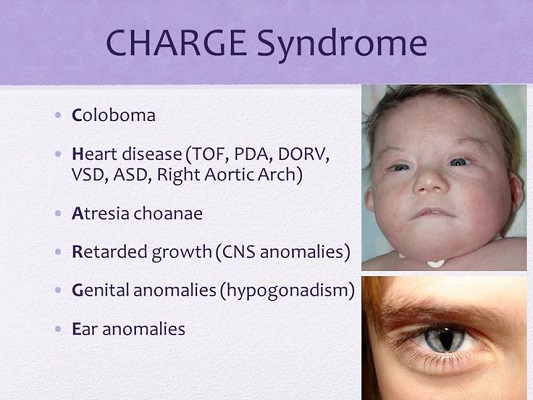
سندرم شارژه یک اختلال نادر است که در طول رشد جنین اولیه ایجاد می شود و بر سیستم های مختلف تأثیر می گذارد . کلمه CHARGE از اولین حرف برخی از ویژگی های رایج تر در کودکان دیده می شود: (C) = کلوبوما (معمولا رتینوکوروئیدال) و نقص عصب جمجمه | (H) = نقص های قلبی ، به خصوص تترالوژی فالوت | (A) = آترزی کوآنا (تنفس بینی مسدود شده) | (R) = عقب ماندگی رشد | (G) = عدم رشد دستگاه های تناسلی | (E) = اختلالات گوش و کاهش شنوایی . همراه کافه پزشکی باشید .
علائم سندرم شارژه از یک کودک به یک دیگر متفاوت است. علت شارژه معمولا جهش (تغییر) جدید در ژن CHD7 یا به ندرت، تغییرات ژنومی در منطقه کروموزوم ۸q12.2 که در آن ژن CHD7 واقع شده است . بازسازی های سه بعدی اسکن MRI نشان دهنده ناهنجاری های استخوانی در بیش از ۸۵٪ بود.
آنچه در این پست کافه پزشکی خواهید دید
علائم و نشانه های سندروم شارژه
سندرم شارژه بر سیستم های مختلف تأثیر می گذارد و منجر به مشکلات متعدد در هنگام تولد می شود. سایر ویژگی های سندروم شارژه ممکن است تا بعدا=ها در زندگی مشخص نشود. تشخیص سندرم شارژه باید توسط یک پزشک ژنتیک بر اساس وجود حداقل یک معیار عمده و چند معیار جزئی سندرم شارژه تشخیص داده شود (نگاه کنید به پایین).
معیارهای عمده تشخیص (۴ C):
این ویژگی ها معمولا در شارژه دیده می شود : کولوباما، اختلالات عصب مغزی، آترتسیس کوآنا، گوش تیپیکال
کولوبوما
کولوبوما یک شکاف در نزدیکی چشم در طول رشد جنین است. این می تواند موجب ایجاد یک سوراخ (کریبوس آریز) و یا اختلالات در شبکیه، ماکولا یا عصب بینایی شود. چشمهای بسیار کوچک (میکروفتالمی) یا بینایی از دست رفته می توانند فرم شدید کلوبوما باشند. کولوباما از شبکیه یا عصب بینایی ممکن است باعث کاهش چشمگیر بینایی، از جمله نقاط کور یا مشکلات درک عمق شود. کلوبوما اغلب در شبکیه رخ می دهد و در حداقل ۸۰-۹۰٪ بیماران مبتلا به سندرم شارژه وجود دارد. کولبومات رتینوکوروئیدی دو طرفه بزرگ ، ویژگی چشمگیری از سندرم شارژه در بیماران مبتلا به جهش CHD7 تایید شده است. جراحی نمی تواند کولوباماهای چشم را اصلاح کند. نزدیکی بینایی یا دوربینی می تواند با عینک کمک کند. عینک آفتابی و کلاه با لایحه محافظتی می توانند کمک کنند.
اختلالات عصب مغزی
کاهش شنوایی حساس به عصب در شارژه ناشی از اختلالات در عصب جمجمه VIII است. از دست دادن شنوایی و دشواری با تعادل، شایع ترین ویژگی های مرتبط با هیپوپلیزی کچلیار و کانال های نیم کره ای است. افت شنوایی می تواند از کاهش شنوایی ملایم تا عمیق باشد. بسیاری از کودکان با شارژه کاشت حلزون را دریافت می کنند تا به حس شنوایی خود کمک کنند.
اکثر کودکان با شارژه دارای مشکلات بلع (اعصاب جمجمه IX / X) هستند. این مشکلات بلع عبارتند از عدم توانایی هماهنگی خوردن و بلعیدن، منجر به عقب رفتن و قرار گرفتن غذا در ریه ها (که می تواند باعث ایجاد پنومونی شود). بسیاری از کودکان نیاز به تغذیه از طریق یک لوله گاستروستومی (لوله به طور مستقیم به معده از طریق دیواره شکمی) تا زمانی که بتوانند با خیال راحت غذا بخورند .
بسیاری از کودکان با شارژه فلج صورت نامتقارن را دارند که منجر به فلج یک طرف چهره (عصب جمجمه VII) می شود. این امر منجر به فقدان بیان صورت می شود که مهم است هنگامی که یک کودک با معلمان یا درمانگران همکاری می کند.
اکثر کودکان با شارژه دارای احساس بوی نامطبوع یا کاهش یافته اند (عصب جمجمه ای است که منجر به نارسایی و کاهش احساس بویایی می شود) . تست بویایی می تواند وجود هیپوگنادیستی هیپوگونادوتروپیک را پیش بینی کند. ترکیبی از التهاب معیوب با هیپوگنادیسم هیپوگنادوتروپیک (نامیده می شود سندرم کالمن) منجر به ایجاد اندام های کوچک خارجی می شود. این در سندرم CHARGE بسیار رایج است و مشورت با یک متخصص غدد درون ریز را ضروری می داند.
آترزی کوآنا
Choanae عبور از پشت بینی به گلو است که امکان نفس کشیدن از طریق بینی را می دهد. در حدود نیمی از تمام کودکان مبتلا به شارژه، این قسمت ها ممکن است مسدود شده باشند (آترزی) یا تنگ (تنگی). جراحی اغلب می تواند این نقص را تصحیح کند . بیماران مبتلا به آترزی یکطرفه معمولا با یک روش جراحی در یک دوره بعد (متوسط ۶ ساله، محدوده ۶ ماه تا ۱۸ سال) تصحیح می شوند، در حالی که بیماران مبتلا به فرم دو طرفه نیاز به مداخله در سن زایمان دارند . اگر هر دو طرف تحت تأثیر قرار گیرند، اقدامات فوری باید انجام شود تا نوزاد بتواند به درستی نفس بکشد و از مشکلات تنفسی جلوگیری کند.
گوش شارژه
اکثر کودکان با شارژه گوش غیر معمولی دارند. گوش تیپیکال شارژه کوتاه و گسترده است . مرکز گوش (concha) اغلب به شکل مثلثی است. گوش اغلب فلاپی است و ممکن است به دلیل غضروف ضعیف باشد. دو گوش اغلب با یکدیگر متفاوتند. یافته های غیرمعمولی در گوش میانی در شارژه نیز وجود دارد، از جمله استخوان های ناقص گوش میانی (۹۳٪) . در بسیاری از موارد، گوش خارجی می تواند به اندازه کافی منحصر به فرد باشد تا مشکوک به شارژه قبل از بررسی ویژگی های دیگر شویم و یک CT scan استخوانی به دنبال کانال های نیم کره ای موجود و ارزیابی کوآنا برای آترزی یا تنگی ضرورت یابد .
علل سندروم شارژه
علت شارژه معمولا جهش (تغییر) جدید در ژن CHD7 یا به ندرت، تغییرات ژنومی در منطقه کروموزوم ۸ (۸q12.2) که در آن ژن CHD7 واقع شده است. عملکرد CHD7 برای توسعه نورون های شبکیه و حرکتی مورد نیاز است. بیش از ۹۰٪ از بیماران معمولی شارژه دارای جهش در ژن CHD7 هستند، در حالی که ۶۵-۷۰٪ از موارد معمول و مشکوک ترکیبی جهش های CHD7 را نشان می دهد. جهش ها به طور مساوی در ناحیه کدگذاری CHD7 توزیع می شوند و اکثر آنها جهش های بی معنی یا تغییر فام هستند. توالی درونی یا پروتئوریک بازآرایی ژنوم پیچیده می تواند از طریق توالی فعلی بدون تکثیر ، حذف یا تجزیه و تحلیل MLPA از دست رفته باشد. اغلب موارد سندرم شارژه به طور پراکنده ای رخ می دهد، اغلب در ارتباط با سن پدر بالاتر . در ۱۲ نفر از ۱۳ خانواده، جهش به آلل پدری (۳/۹۲٪) و میانگین سن پدر در هنگام تولد ۳۳ سال منجر شد. در موارد نادر، شارژه در خانواده ها، یا دو کودک مبتلا به بیماری یا یک والد و کودک تحت تاثیر قرار گرفته است، یا به دلیل موزاییکایی والدین برای جهش CHD7، که باعث می شود که والدین به طور ضعیف تحت تاثیر قرار می گیرند یا در همه آنها تاثیر نمی گذارد.
هیچ تراتوژن شناخته شده ای (در معرض حاملگی) وجود ندارد که با سندرم شارژه مرتبط باشد. اسید رتینوئیک یا ایزوترتینین (داروهایی که برای آکنه شدید مصرف می شوند) در طول سه ماهه اول بارداری ممکن است باعث ناهنجاری های مشابهی شوند.
تشخیص سندروم شارژه
یک ژنتیک پزشکی یا متخصص دیگری که با سندروم شارژه آشنا است، باید یک امتحان کامل و یک آزمایش کامل را انجام دهد تا ویژگی های اصلی و جزئی شارژه که در بالا ذکر شده است را جستجو کند. سایر اختلالات مشابه مانند سندرم حذف ۲۲q11.2، سندرم مولات ویلسون، سندرم کوبوکی، سندرم کالمن و ضعف عصبی EFTUD2 (سندرم نقص مادرزادی مادرزادی ، اختلال نعوظ چندگانه با تشکیل دیستوستیز مینیبولا فاسیال با آسیب ناحیه گوش خارجی، افت شنوایی، شکاف کام ، آترزی کوآنا، میکروسفالی، اختلال ناتوانی، آترزی مری، نقص مادرزادی قلب و نقص اشعه رادیویی) نیز باید کنار گذاشته شود.
تست ژنتیک مولکولی برای جهش در ژن CHD7 در ارتباط با این بیماری وجود دارد و اگر این منفی باشد، باید یک میکروآرایه کروموزومی SNP انجام شود، زیرا در چند مورد، تغییرات ژنومی در زیر میکروسکوپیک کروموزوم ۸q12.2 وجود دارد. اگر هر دو این آزمایش منفی باشند، باید توالی کامل ژنوم exome را انجام داد، از آنجا که سایر اختلالات ژنتیکی برخی از ویژگی های بالینی را با سندرم شارژه به اشتراک می گذارند، و جهش های de novo در ZEB2، KMT2D و EFTUD2 در کودکان پیش از تشخیص سندرم شارژه تشخیص داده شده است.
درمان سندروم شارژه
اگر چه این کودکان دارای مشکلات فراوانی هستند، آنها می توانند زنده بمانند و سالم و شاد باشند. بسیاری از اختلالات ساختاری (درد شکمی، شکاف لب، و غیره) می توانند با جراحی اصلاح شوند. دیگر مشکلات ، مانند مشکلات تغذیه ای و زبان ممکن است سال ها طول بکشد و سایر مداخلات را به وجود آورد. نوزادان مبتلا به سندرم شارژه باید به دنبال تعدادی از متخصصان پزشکی بسته به نیازهای فردی آنها باشد. بعضی از متخصصان پزشکی که اغلب کودکان مبتلا به سندرم شارژه را دنبال می کنند، شامل ژنتیک، قلب و عروق، گوش و حلق و بینی، چشم پزشکی، اورولوژی و غدد درون ریز هستند.
بیش از ۵۰٪ از کودکان مبتلا به سندرم شارژه اختلالات خواب را تجربه می کنند و آپنه انسدادی خواب در کودکان مبتلا به سندرم شارژه شایع است. تمام درمان های معمول برای آپنه انسدادی خواب باعث کاهش علائم می شود. سم بوتولینوم A (بوتاکس) برای کاهش ترشحات بیش از حد بزاق در یک نوزاد مبتلا به تهویه با سندرم شارژه که نیاز به تراکوتومی دارد استفاده شده است.
در میان ۲۰۲ بیمار مبتلا به جهش CHD7 و سندرم شارژه، طیف گسترده ای از نقص های قلبی در ۷۴٪ از این گروه از بیماران. نقصهای کانوتوریل و نقصهای سپتوم اتریوواستریکال در بیماران مبتلا به جهش CHD7 در مقایسه با بیماران مبتلا به نارسایی قلبی غیر سندرومیک بیش از حد نشان داده شده است. با کار با مدل های ماوس نشان می دهد که CHD7 نقش مهمی در مزودرم قارچی در طی رشد قلب و عروق دارد.
منبع : rarediseases.org | ترجمه کافه پزشکی